本学大学院医歯学総合研究科 脳神経病学・老年医学分野の髙嶋 博教授、樋口雄二郎医師らの研究グループは、東京大学大学院医学系研究科 分子神経学講座 (辻省次 教授)、京都工芸繊維大学・応用生物学系・染色体工学研究室 昆虫先端研究推進センター (山口政光 教授)らとの共同研究により、神経難病「遺伝性末梢神経障害[※1]と脊髄小脳変性症[※2]」の原因遺伝子を発見しました。
ミトコンドリア呼吸鎖複合体アセンブリ(集合)因子をエンコードするCOA7遺伝子の変異が、軸索型ニューロパチーや小脳失調症などの2つの異なる神経難病に特徴的な症状を同時に引き起こすことを突き止め、新しい疾患概念として“軸索型ニューロパチーを伴う脊髄小脳失調症(Spino-cerebellar Ataxia with Axonal Neuropathy type 3”(SCAN3)と名づけました。
遺伝性末梢神経障害と脊髄小脳変性症はいずれも、その病態や遺伝的原因が未解明の部分が多く、根本的治療法の確立されていない神経難病でありますが、本研究によりミトコンドリア機能異常が末梢神経および中枢神経の両方に神経変性を引き起こすことが解明されたことで2つの神経難病の病態理解が深まりました。さらに、本研究では世界で初めてSCAN3のショウジョウバエ疾患モデルの樹立にも成功しました。本研究は、SCAN3のみならず、遺伝性末梢神経障害や脊髄小脳変性症、その他の複数の系統が障害される神経変性疾患のさらなる病態解明や治療開発にも大きく貢献しうる成果です。
本研究の成果は、神経学分野において最も権威のある雑誌の1つである英国神経学雑誌『Brain(ブレイン)』に2018年4月27日に掲載されました。
※1~3の用語解説は、以下の詳細内容に記載しています。
(研究内容に関する問い合わせ先)
鹿児島大学大学院医歯学総合研究科神経病学講座 脳神経内科・老年病学 教授
髙嶋 博 (タカシマ ヒロシ)
〒890-8520 鹿児島県鹿児島市桜ヶ丘8-35-1
Tel: 099-275-5332 Fax: 099-265-7164
Mail: このメールアドレスはスパムボットから保護されています。閲覧するにはJavaScriptを有効にする必要があります。
当科の名称は、4月1日より医歯学総合研究科は脳神経内科・老年病学になっております。
異なる2つの神経難病「遺伝性末梢神経障害と脊髄小脳変性症」を同時に引き起こす新しい原因遺伝子を発見
-病態解明・治療開発に向けての新たな一歩-
【研究の背景】
シャルコー・マリー・トゥース病(Charcot-Marie-Tooth disease; CMT)に代表される遺伝性末梢神経障害は、末梢神経の異常によって四肢の感覚と運動が徐々に障害され、手足が痩せ細り進行すれば歩行も出来なくなる病気です。一方、脊髄小脳変性症は小脳性の運動失調症状を主体とするもので、主に小脳や脳幹の神経細胞が選択的に変性、脱落する病気です。いずれも遺伝性の神経難病であり、多くの原因遺伝子が明らかとなっていますが、その病態や発症のメカニズムは未解明の部分が多く、未知の原因遺伝子があると考えられていました。いずれの神経難病も病態の解明や治療法の確立は今後の大きな課題であり、そのためには原因の解明が必要不可欠であります。
【研究の内容】
鹿児島大学脳神経内科では、2007年以降、全国各地の医療機関から遺伝性末梢神経障害や脊髄小脳変性症の遺伝子診断の依頼を受けており、これまでに2000例を超える患者の遺伝子解析を継続して実施しておりました。しかし遺伝子異常が見つかる患者は半数以下であり、未知の原因遺伝子が多く存在すると考えられていました。本研究は、新たな原因遺伝子の探索するため、原因未同定の遺伝性末梢神経障害の患者を対象として、次世代ゲノムシークエンサーを用い、ヒト遺伝子のほぼすべてを網羅的に解析するエクソーム解析[※3]を東京大学と共同で行いました。
多数例の遺伝子変異の解析は膨大なものでありましたが、その中から鹿児島大学の樋口雄二郎医師は、患者4例に共通してCOA7遺伝子の異常を有していることを発見し、さらにこの4症例の臨床像が末梢神経障害のみならず脊髄小脳変性症の特徴を有していることも見いだしました。全例とも発症年齢が幼少期と比較的早く、両下肢遠位部の筋力低下、筋萎縮、感覚障害に加え小脳性運動失調を呈していることが共通していました。特徴的な臨床症状を呈していたことから、COA7遺伝子異常に引き起こされる疾患を『軸索型ニューロパチーを伴う脊髄小脳変性症(Spino-cerebellar Ataxia with Axonal Neuropathy type 3: SCAN3)』と命名しました。
COA7遺伝子から作られるシトクロムcオキシダーゼ・アセンブリ因子(Cytochrome C Oxidase Assembly Factor 7)というタンパク質は、様々な組織の細胞のミトコンドリア内に豊富に発現しており、呼吸鎖複合体のアセンブリ(集合)に重要な役割をもつことが知られていました。本研究では、COA7が末梢神経および中枢神経系に豊富に発現していることが明らかにし、COA7変異をもつ患者のMRI検査にて小脳や脊髄に萎縮をきたしていることや大脳白質に病変を呈していることを見いだしました(図1)。また患者から採取した末梢神経組織にて軸索障害をきたしていること、筋組織にてミトコンドリア異常をきたしていること(図1)、皮膚組織から培養した線維芽細胞を用いて、ミトコンドリア呼吸鎖複合体の酵素活性が非常に低下していることを明らかにしました。さらに、本研究では京都工芸繊維大学昆虫先端研究推進センター 山口政光 教授のもとCOA7をノックダウンしたショウジョウバエの疾患モデルの樹立に正解で初めて成功し、複眼の形態異常や運動機能の低下、寿命の短縮、神経筋接合部のシナプス形態異常が誘導されることを明らかにし、COA7の機能障害による神経変性のメカニズムの一部を解明しました(図2)。
本研究は、遺伝性末梢神経障害や脊髄小脳変性症を含むさまざまな神経変性疾患に共通する神経変性メカニズムの解明および今後の治療開発にも大きく貢献しうる成果です。さらに本研究で樹立したショウジョウバエ疾患モデルを用いることで、今後は薬剤スクリーニング系を用いた治療法開発を期待できます。 本研究の成果は、神経学分野において最も権威のある雑誌の1つである英国神経学雑誌『Brain(ブレイン)』に2018年4月24日に掲載されます。
【用語解説】
※1 遺伝性末梢神経障害:遺伝的原因により末梢神経(運動または感覚神経)の障害が引き起こされる疾患
の総称であり、その代表としてシャルコー・マリー・トゥース病があげられる。臨床的には四肢遠位筋優
位の進行性筋力低下や逆シャンペンボトル様下腿筋萎縮、凹足、手袋靴下型の感覚障害、深部腱反射の減
弱などを特徴とする。発症年齢は生下時~高齢と広範であるが、重症例では寝たきりや嚥下障害や呼吸筋
麻痺を来たし人工呼吸器を必要とする症例もまれにいる。
※2 脊髄小脳変性症(spinocerebellar degeneration; SCD):SCDは歩行時のふらつきや、手の震え、ろれ
つが回らない等を症状とする神経の病気であり、主に小脳という部分の働きが悪くなる神経変性疾患の総
称である。根本的な治療法のない難病であり、発症年齢は生下時~高齢と広範であるが、重症例では寝た
きりになる症例もいる。約1/3に遺伝歴を認め、遺伝性のものは常染色体優性遺伝および劣性遺伝、X連
鎖性のものが知られている。
※3 エクソーム解析:約2.3万個のヒト遺伝子のエクソン領域のみを網羅的に解析する手法。エクソン領域
は全ゲノムの約1~1.5%に過ぎないが、タンパク質に翻訳される領域であることから機能的に重要であ
り、遺伝性疾患の多くがエクソン領域の変異により引き起こされると推定されている。
※4 エンコード:遺伝子が、あるタンパク質を作るためのアミノ酸配列に対する遺伝情報を持っているこ
と。
【発表雑誌】
掲載雑誌:Brain (https://academic.oup.com/brain)
論文名:Mutations in COA7 cause spinocerebellar ataxia with axonal neuropathy
著者名:Yujiro Higuchi, Ryuta Okunushi, Taichi Hara, Akihiro Hashiguchi, Junhui Yuan, Akiko Yoshimura,
Kei Murayama, Akira Ohtake, Masahiro Ando, Yu Hiramatsu, Satoshi Ishihara, Hajime Tanabe,
Yuji Okamoto, Eiji Matsuura, Takehiro Ueda, Tatsushi Toda, Sumimasa Yamashita,
Kenichiro Yamada, Takashi Koide, Hiroaki Yaguchi, Jun Mitsui, Hiroyuki Ishiura, Jun Yoshimura,
Koichiro Doi, Shinichi Morishita, Ken Sato, Masanori Nakagawa, Masamitsu Yamaguchi,
Shoji Tsuji, Hiroshi Takashima
本研究は、「次世代遺伝子解析技術を用いた希少難治性疾患の原因究明及び病態解明に関する研究」(厚生労働省)および日本医療研究開発研究機構(AMED)による支援を得て行われました。 本研究にご協力頂きました患者様とご家族の皆様に深謝致します。
【参考資料】
図1:COA7変異を有するSCAN3患者の臨床的および組織学的特徴
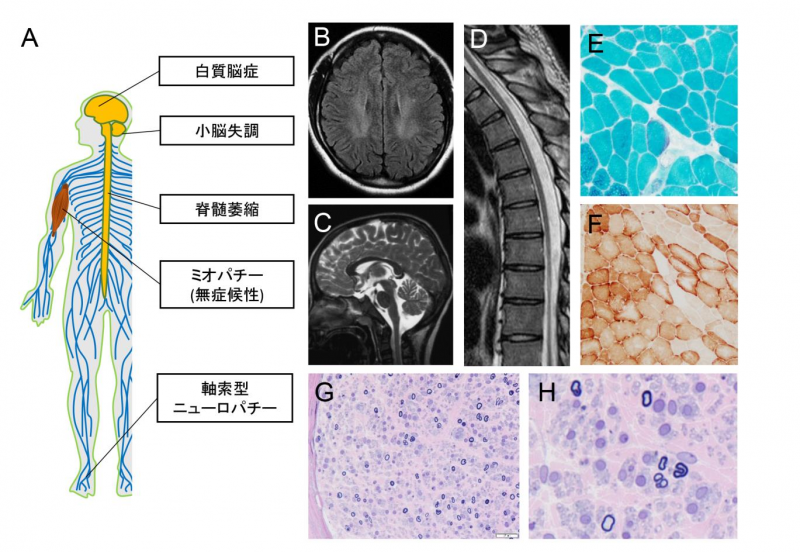
COA7変異を有するSCAN3患者は白質脳症や小脳失調、脊髄萎縮、無症候性ミオパチー、軸索型ニューロパチーを呈する(A)。患者の頭部MRIでは白質脳症(B)、小脳および脊髄の萎縮(C、D)を認める。筋生検ではミトコンドリア異常を示唆する赤色ぼろ線維(E)およびCCO欠損線維(F)を認め、神経生検では大径有髄線維密度の高度の低下を認める(G、H)。
図2:SCAN3のショウジョウバエ疾患モデル
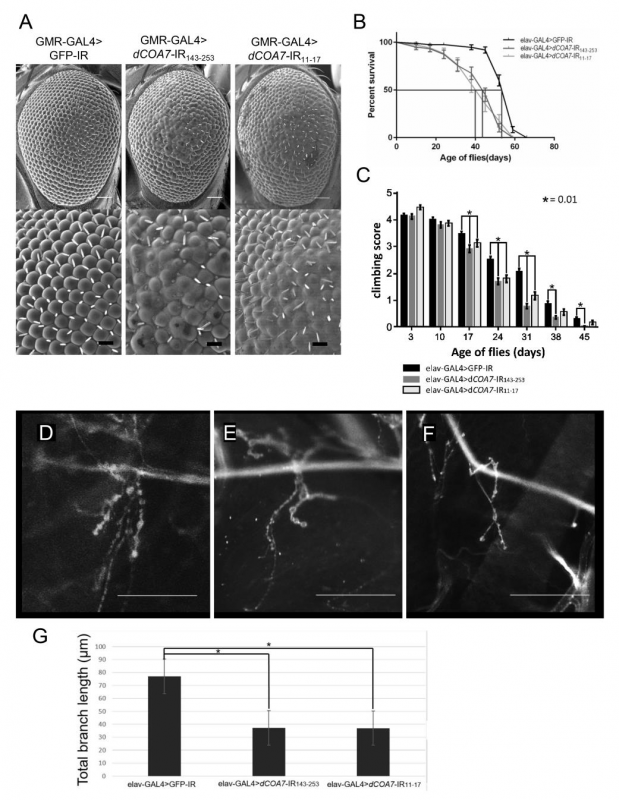
COA7をノックダウンしたショウジョウバエでは、複眼の形態異常(A)や寿命の短縮(B)、運動機能の低下(C)、神経筋接合部のシナプス形態異常が誘導されることが明らかになった(D-G)。













